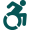
We have access to two massive parallel computer clusters, and to a few smaller clusters. These include 192 Intel Xeon processor cluster of the Institute of Physics of the MCSU, and 1552 Intel Xeon processor cluster of the Faculty of Mathematics, Physics and Computer Science of the MCSU. Both DFT codes, the SIESTA and VASP, allow to take the advantage of full available computing resources. Thus calculations taking into account huge systems, containing even a thousand of atoms, are possible.
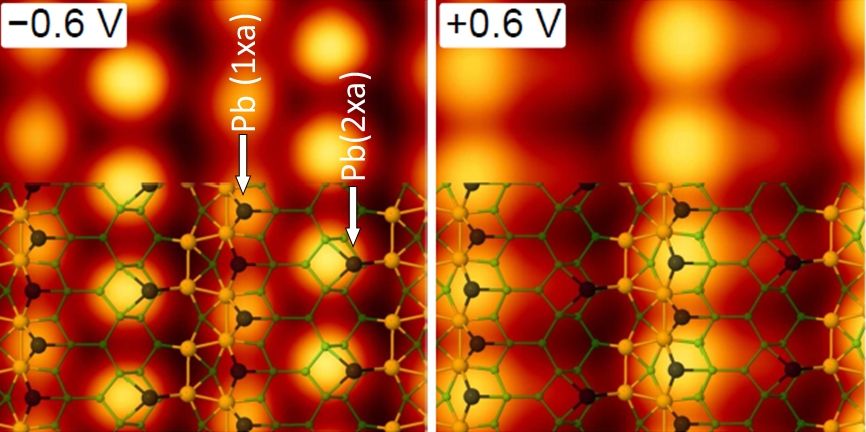
Both DFT codes give a possibility to relax atomic positions, find a minimum energy structural model of the system and study its structural and electronic properties. Considering different initial positions of atoms, we are looking for the lowest energy structure in a given supercell geometry. The resulting geometry can be compared with the RHEED data. Once the structure is found, the bonding of the atoms can be studied. This gives us the information where and why the atoms bond. The STM topography simulations within the Tersoff-Hamman approach can also be performed and compared to the experimental STM data.
Both codes have the capability of performing STM simulations for various bias voltages and tunneling currents, thus allowing for a direct comparison with the experiment. This also concerns the electronic properties, in particular density of states of the surface structure, which can be verified by the STS measurements.
Finally, the electronic band structure of the system under consideration can also be obtained. The VASP code can perform the calculations taking into account effect of spin-orbit interaction. Again, theoretical results can be compared with the (spin-polarized) ARPES data. Furthermore, it is also possible to determine the character and origin of the bands, which is very important in understanding of the properties of the nanostructures and its interaction with the substrate.
Theoretical calculations within the tight binding model allow us to analyze the conductance, charge and density of states of atomic chains and atom clusters on different surfaces. In particular we concentrate on the role of electron localization/delocalization in the substrate on the surface transport properties.